Instead of heading towards the more theoretic graph design, the day after writing part 1 of what is turning out to be a series, I focused on concrete software changes that might answer the first question I posed in the previous post (“If you recast the problem as that of calling ‘annotated variants’, can you speed up the current pipelines?”), and that is what this post will be about. The more theoretic graph design will be coming, but I’m still thinking through Heng Li’s update describing previous work on graph algorithms, as well as a recent slide deck he presented (the picture of where he is coming from is becoming clearer…).
I also decided to do something “foolish” for this part of the work. On a regular basis, people lament about wanting better bioinformatics tools, and about how all they get are new tools (most of which may or may not be better, because they have only been run on the datasets needed to publish the papers). Which leads others to reiterate all of the reasons why academic bioinformatics software is written that way (papers & citations all important, difficulty publishing updated software, no funding for maintenance or improvement).
All of which is true, and is my situation right now, but since I’m still in my honeymoon period here at Yale, let’s go make some “better” bioinformatics tools and see what happens.
The Results
The tool I chose is (no surprise) BWA MEM, and to pique your interest (or really, just to have you not stop reading when I talk about all that boring algorithm/software stuff), I’ll start with the results. I took the current development head of BWA, made the changes described below, and then ran it on the NA12878 exome and genome datasets also used by Blue Collar Bioinformatics in comparing variant calling pipelines.
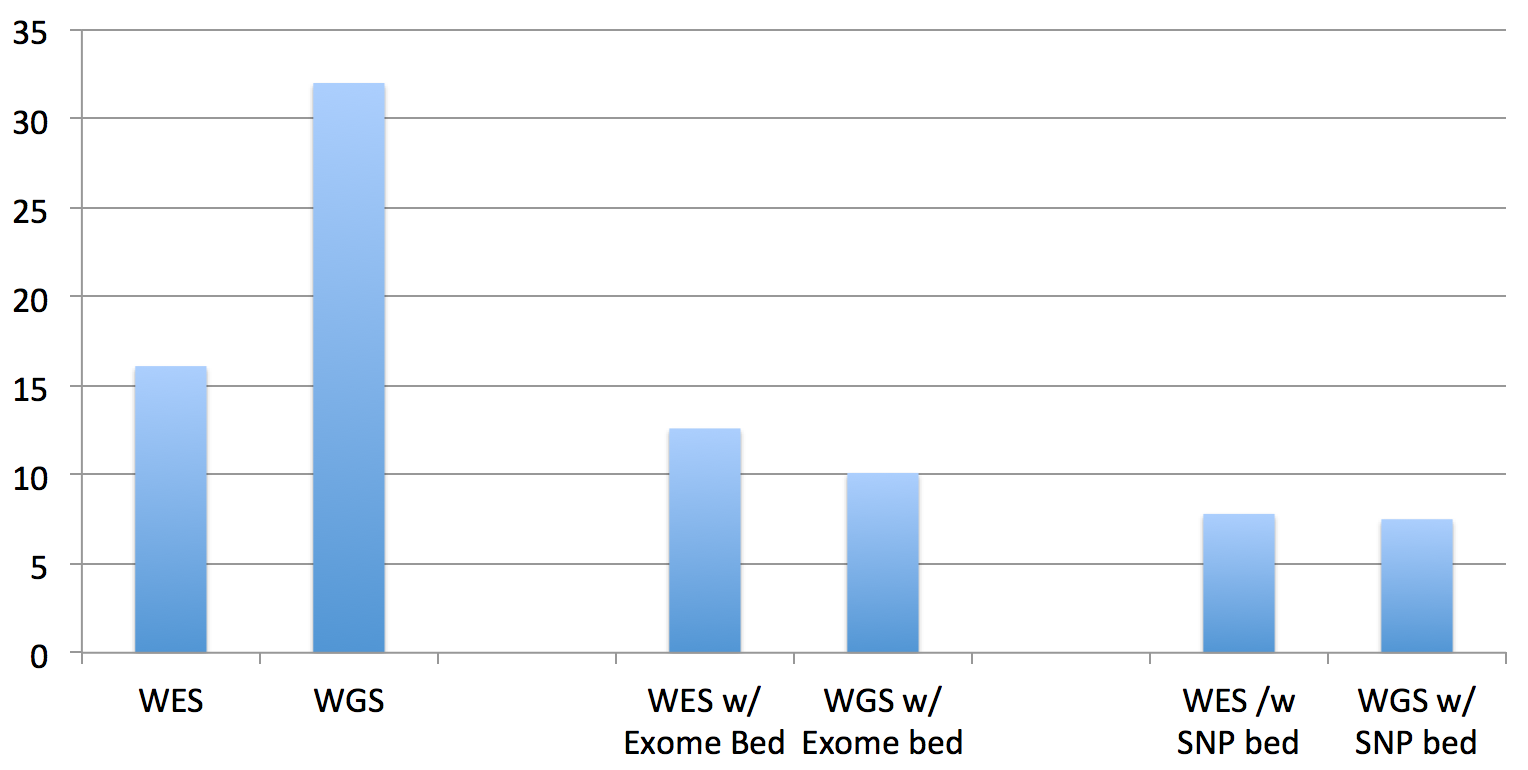
The first two columns are the “run times” of the standard bwa mem on the exome and genome data. The middle two columns are the “run times” of the updated BWA, given the exome’s BED file of target regions. The right two columns are the “run times” of the updated BWA, given a BED file of all of the “flagged” SNPs in dbSNP138 (these are the clinically actionable variants).
And, by “run time”, what I’m reporting here are the interval times that BWA MEM reports as it processes each batch of reads, which is very consistent across the batches. The total running time is linear in this interval time and the size of the datasets (since I’m still in development mode, as you’ll see below, quick but representative times work better…when we get to done, I’ll do timings and results for the whole process).
So, the total running times are not quite as significant as it appears in that chart for WGS datasets, since you have to multiply by the difference in the data size (the recent 30x HiSeq X Ten dataset for NA12878 dataset is approx. 10 times larger than this exome dataset):
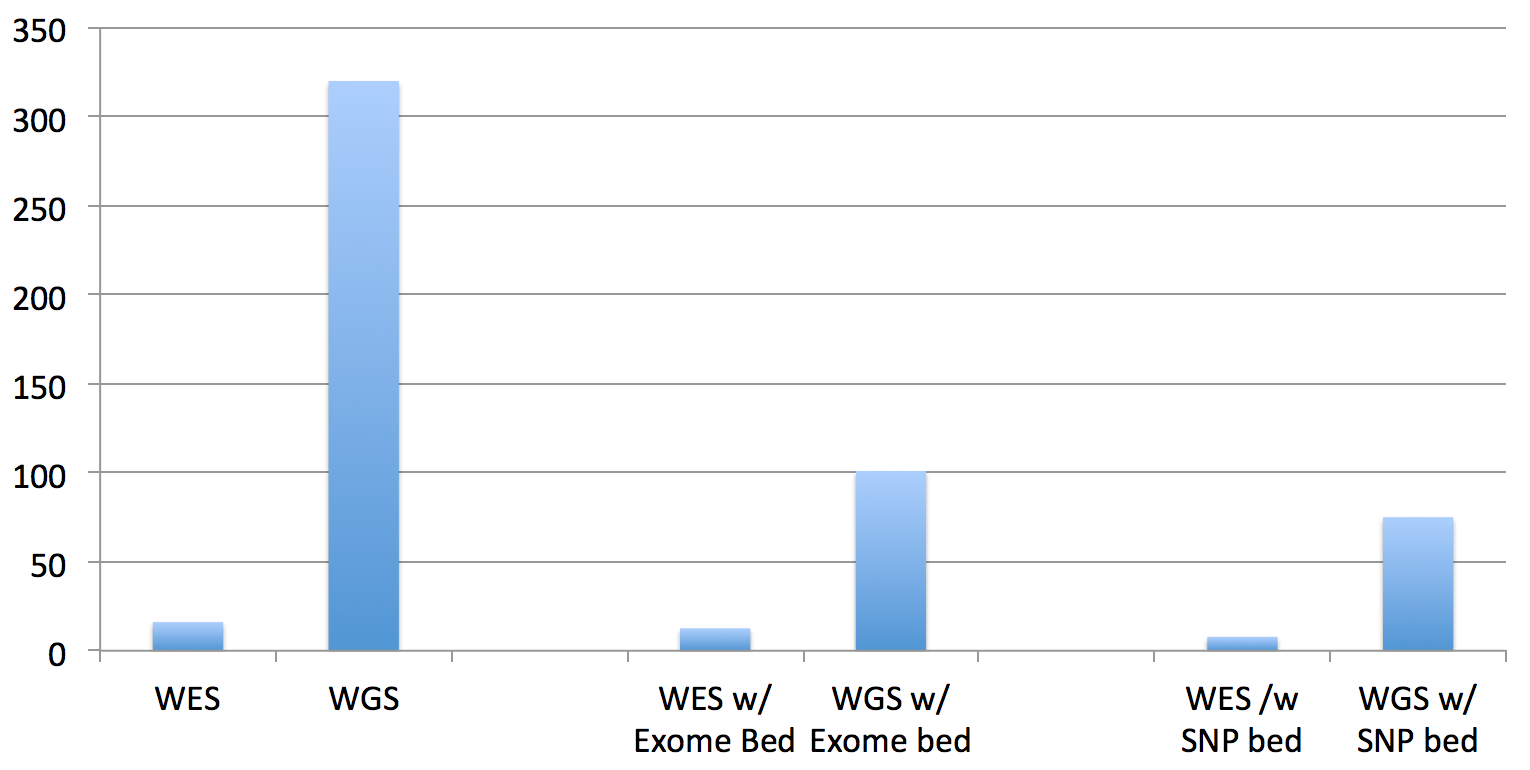
An improvement for both exomes and genomes, yes, but can we do better? The answer to that is also yes, I believe, but it will take answering the questions at the end of the post and the completion of the software development.
The Methods
The set of changes to bwa were on the simpler side, without any substantial algorithmic changes. An option was added to bwa mem to pass a bed file of target intervals to the algorithms, and then checks were added to stop the alignment process when a read will not align in one of those intervals.
The given intervals are expanded by 300 bp on each side, to ensure that there are few to no boundary effects for the paired-end reads mapping to the actual intervals. The data structure used for doing the interval checks is discussed in the next section, because it may also be used to speed up the variant calling process later in the pipeline.
In the main algorithm, checks were added at the following points (if you don’t know what SMEMs, MEMs or chains are, either read the BWA and BWA MEM papers…or just think of SMEMs and MEMs as the initial exact matches and chains as the larger, potentially gapped, matches):
- At the first SMEM, if it covers the entire trimmed region (which means that there is an exact match to the “whole” read)
- After the generation of MEMs
- After the generation of chains
- After the generation of the final alignment
Note: For the first check to be effective, bases at the 5’ and 3’ ends with quality score less than 5 are “trimmed” for the identification of SMEMs and MEMs (the full read is used for the rest of the algorithm and in the output). Without this trimming step, the test to see if the whole read exactly matches the reference rarely succeeds, and the overall run time doubles.
A read fails the check if all of the SMEMs, MEMs, chains or alignments are outside the target intervals, the computation stops for that read at that point, and no output is generated for that read. Also, reads are dropped from the output if either they are unaligned or the pair has a mapping quality score of 0.
Note: This last condition is a long-standing belief on my part that true repeat reads have no place in a variant calling application, because either the read exactly matches the multiple genome locations (in which case, there are no variants), or any sequence difference that might lead to a variant call cannot be attributed to a specific genome location, and so the resulting variant is likely to be an error. And, the Erik Garrison (lead author of freebayes) notes near the bottom of a response about trimming recommendations, he is currently filtering out these pairs in the pipeline he runs. But, I would like to hear from someone who does use these reads in variant calling.
Next Steps
Okay, so we’ve gotten a decent speedup of the algorithm for variant calling of exome regions, disease gene panels or local regions like the clinically actionable variants. But, can it be better?
The reason why I think it could is that there is now a hotspot in the computation, specifically at the calculation of the initial SMEM for each read. The “run time” when you just short circuit the computation just before that step is ~1 (instead of 7-10 when given BED files). So, it looks like up to 50-70% of the current computation could be eliminated for WGS datasets, if we can make a very quick check for off-target reads.
My thought is whether you can flag the sections of the BWT index to signal that a read is aligning somewhere else in the genome, and short-circuit the SMEM computation at that point. Or, can you construct another data structure that can quickly determine that a read does not align to any of the target intervals, without computing the full SMEM? I believe the answer to that is yes, but it is not clear to me how to do that now. Any ideas from the BWT algorithm experts? If you haven’t seen it yet, the methods described above, and the answer to this question, are applicable to any of the current mapping tools (Bowtie, MOSAIK, Novoalign, …) and should provide a similar speedup.
Moving over to the other major computation, variant calling. (With this solution, the sorting, duplicate removal and indexing steps for WGS datasets will be on par with WES datasets, since only the reads aligning in the target intervals make it into the BAM file.) My thought here is that, for exomes, the caller computes over 40-60 million bases of the reference, but only ends up with 40-50 thousand variants. Can we, prior to variant calling, subset the target intervals to only those that have enough reads that differ from the reference, and just give the variant caller that subset?
I’m of two minds about how to do that. Initially, my thought was to build it into the aligner, because we are seeing all of the reads and their alignments, so without any additional I/O, we can piggyback this computation to it. Hence, the data structure I’m using right now to mark the target intervals in the modified bwa is an array of uint8_t’s, for each base of the reference, and the software parses the cigar strings of the output read alignments, and counts any differences at each reference position. This way, you can then scan the counts to determine which subset of regions have enough differences to make it worthwhile to use the variant caller on it (and more importantly, skip the regions without enough differences).
Note: Yes, I know that a 1-byte counter for each reference position is 3 gigabytes of memory for human references, and is overkill. The real data structure will be much more efficient, once we figure out what the actual algorithm is going to be. (Initally, I partitioned the reference into 10 base windows, and kept counts for each window. But, when I started thinking about how to compute the difference regions, the window-based counts didn’t fit well.)
My current thought is to run a 10bp sliding window to identify those windows with either > 1 difference at a reference position or more than 6 columns with 1 difference. But, these are preliminary thoughts right now, since the focus was on the alignment step.
But, as I started writing this last section, I couldn’t come up with any reason why this target interval recalculation can’t be a separate step of the pipeline as a whole (possibly between the duplicate removal and the variant calling), creating a trimmed interval list to give to the variant caller. Scanning a BAM file once is not that computationally intensive. Has anyone done one of those, or does the variant calling algorithms already do that kind of filtering right now? This is the next speedup step after the alignment computation has been optimized.
And then, finally, after the above implementations are complete, we need to check the alignments in the target intervals and the final variant calls to make sure we’re not dropping any relevant reads or calls because of the speedups. The next post on the software improvements will report on all of this.
But, I think my next post in the series will turn back to the graph design question, and think through in detail what the graph format needs to supply to the downstream tools (and so what needs to be in there). Until then..